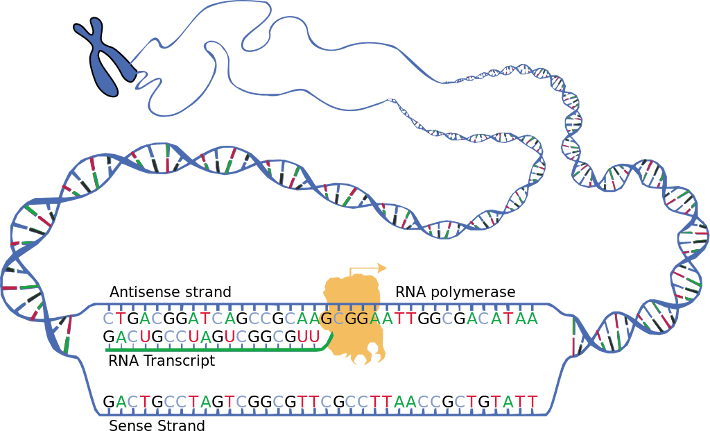
Das Transkriptom ist die Menge aller RNA-Transkripte, einschließlich kodierender und nicht-kodierender, in einem Individuum oder einer Zellpopulation. Der Begriff kann sich manchmal auch auf alle RNAs oder nur auf mRNA beziehen, je nach dem jeweiligen Experiment
DDas Transkriptom ist die Menge aller RNA-Transkripte, einschließlich kodierender und nicht-kodierender, in einem Individuum oder einer Zellpopulation. Der Begriff kann sich manchmal auch auf alle RNAs oder nur auf mRNA beziehen, je nach dem jeweiligen Experiment. Der Begriff Transkriptom ist ein Portmanteau aus den Wörtern Transkript und Genom; er bezieht sich auf den Prozess der Transkriptproduktion während des biologischen Prozesses der Transkription.
Die Anfänge der Transkriptom-Annotation begannen mit cDNA-Bibliotheken, die in den 1980er Jahren veröffentlicht wurden. Später führte das Aufkommen der Hochdurchsatztechnologie zu schnelleren und effizienteren Möglichkeiten, Daten über das Transkriptom zu erhalten. Zwei biologische Techniken werden zur Untersuchung des Transkriptoms verwendet, nämlich DNA-Microarray, eine auf Hybridisierung basierende Technik, und RNA-seq, ein sequenzbasierter Ansatz.1Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660. RNA-seq ist die bevorzugte Methode und ist seit den 2010er Jahren die dominierende Transkriptomik-Technik. Die Einzelzell-Transkriptomik ermöglicht die Verfolgung von Transkriptveränderungen über die Zeit innerhalb einzelner Zellen.
Die aus dem Transkriptom gewonnenen Daten werden in der Forschung genutzt, um unter anderem Einblicke in Prozesse wie zelluläre Differenzierung, Karzinogenese, Transkriptionsregulation und Biomarker-Entdeckung zu gewinnen. Transkriptom gewonnene Daten finden auch Anwendung bei der Erstellung phylogenetischer Beziehungen während des Evolutionsprozesses und bei der In-vitro-Fertilisation. Das Transkriptom ist eng verwandt mit anderen –om-basierten biologischen Untersuchungsfeldern; es ist komplementär zum Proteom und zum Metabolom und umfasst das Translatom, Exom, Meiom und Thanatotranskriptom, die als ome Bereiche angesehen werden können, die spezifische Arten von RNA-Transkripten untersuchen. Es gibt zahlreiche öffentlich zugängliche Transkriptom-Datenbanken.
Etymologie und Geschichte
Das Wort Transkriptom ist ein Portmanteau aus den Wörtern Transkript und Genom. Es erschien zusammen mit anderen Neologismen, die mit den Suffixen -ome und -omics gebildet wurden, um alle Studien zu bezeichnen, die auf genomweiter Ebene in den Bereichen Biowissenschaften und Technologie durchgeführt werden. Als solche waren Transkriptom und Transkriptomik neben Genom und Proteom eines der ersten Wörter, die auftauchten.2 Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020. Die erste Studie, die einen Fall einer Sammlung einer cDNA-Bibliothek für Seidenspinner-mRNA vorstellte, wurde 1979 veröffentlicht.3GK, Sim; FC, Kafatos; CW, Jones; MD, Koehler; A, Efstratiadis; T., Maniatis (December 1979). “Use of a cDNA library for studies on evolution and developmental expression of the chorion multigene families”. Cell. 8 (4): 1303–16. doi:10.1016/0092-8674(79)90241-1. PMID 519770. Die erste bahnbrechende Studie, die das Transkriptom eines Organismus erwähnte und untersuchte, wurde 1997 veröffentlicht und beschrieb 60.633 Transkripte, die in S. cerevisiae mittels serieller Analyse der Genexpression (SAGE) exprimiert wurden. Mit dem Aufkommen der Hochdurchsatztechnologien und der Bioinformatik und der damit einhergehenden gesteigerten Rechenleistung wurde es immer effizienter und einfacher, enorme Datenmengen zu charakterisieren und zu analysieren.4Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020. Die Versuche, das Transkriptom zu charakterisieren, wurden mit dem Aufkommen der automatisierten DNA-Sequenzierung in den 1980er Jahren stärker. In den 1990er Jahren wurde die Expressed-Sequence-Tag-Sequenzierung zur Identifizierung von Genen und deren Fragmenten eingesetzt5Govindarajan, Rajeshwar; Duraiyan, Jeyapradha; Kaliyappan, Karunakaran; Palanisamy, Murugesan (2012). “Microarray and its applications”. Journal of Pharmacy and Bioallied Sciences. 4 (6): S310-2. doi:10.4103/0975-7406.100283. PMC 3467903. PMID 23066278., gefolgt von Techniken wie der seriellen Analyse der Genexpression (SAGE), der Kappenanalyse der Genexpression (CAGE) und der massiven parallelen Signatursequenzierung (MPSS).
Transkription
Siehe auch: Transkription (Biologie)
Das Transkriptom umfasst alle Ribonukleinsäure (RNA)-Transkripte, die in einem bestimmten Organismus oder einer experimentellen Probe vorhanden sind.6C Frith, Martin; Pheasant, Michael; S Mattick, John (2005). “Genomics: The amazing complexity of the human transcriptome”. European Journal of Human Genetics. 13 (8): 894–897. doi:10.1038/sj.ejhg.5201459. PMID 15970949. S2CID 2836126. RNA ist der Hauptträger der genetischen Information, der für den Prozess der Umwandlung der DNA in den Phänotyp eines Organismus verantwortlich ist. Ein Gen kann durch einen molekularen Prozess, der als Transkription bekannt ist, zu einer einzelsträngigen Boten-RNA (mRNA) führen; diese mRNA ist komplementär zu dem DNA-Strang, aus dem sie entstanden ist.7Peralta, Mihaela (2012). “The Human Transcriptome: An Unfinished Story”. Genes. 3 (3): 344–360. doi:10.3390/genes3030344. PMC 3422666. PMID 22916334. Das Enzym RNA-Polymerase II heftet sich an den Template-DNA-Strang und katalysiert die Addition von Ribonukleotiden an das 3′-Ende der wachsenden Sequenz des mRNA-Transkripts.8Clancy, Suzanne (2008). “DNA Transcription”. Nature Education. 1 (11): 41.
Um ihre Funktion zu initiieren, muss die RNA-Polymerase II eine Promotorsequenz erkennen, die sich stromaufwärts (5′) des Gens befindet. In Eukaryoten wird dieser Prozess durch Transkriptionsfaktoren vermittelt, insbesondere durch den Transkriptionsfaktor II D (TFIID), der die TATA-Box erkennt und die Positionierung der RNA-Polymerase an der entsprechenden Startstelle unterstützt. Um die Produktion des RNA-Transkripts zu beenden, findet die Terminierung statt, die in der Regel mehrere hundert Nuklekotide von der Terminationssequenz entfernt ist, und es kommt zur Spaltung.9Clancy, Suzanne (2008). “DNA Transcription”. Nature Education. 1 (11): 41. Dieser Prozess findet im Zellkern zusammen mit der RNA-Prozessierung statt, bei der mRNA-Moleküle verkappt, gespleißt und polyadenyliert werden, um ihre Stabilität zu erhöhen, bevor sie anschließend ins Zytoplasma gelangen. Aus der mRNA werden durch den Prozess der Translation, der in Ribosomen stattfindet, Proteine gebildet.
Arten von RNA-Transkripten
Gemäß dem zentralen Dogma der Molekularbiologie umfasste das Transkriptom zunächst nur proteinkodierende mRNA-Transkripte. Dennoch existieren mehrere RNA-Subtypen mit unterschiedlichen Funktionen. Viele RNA-Transkripte kodieren nicht für Protein oder haben unterschiedliche regulatorische Funktionen im Prozess der Gentranskription und Translation. RNA-Typen, die nicht in den Geltungsbereich des zentralen Dogmas der Molekularbiologie fallen, sind nicht-kodierende RNAs, die in zwei Gruppen unterteilt werden können: lange nicht-kodierende RNA und kurze nicht-kodierende RNA.
Zur langen nicht-kodierenden RNA gehören alle nicht-kodierenden RNA-Transkripte, die mehr als 200 Nukleotide lang sind. Mitglieder dieser Gruppe machen den größten Anteil des nicht-kodierenden Transkriptoms aus. Kurze nicht-kodierende RNA umfasst die folgenden Mitglieder:
Transfer-RNA (tRNA)
Mikro-RNA (miRNA): 19-24 Nukleotide (nt) lang. Mikro-RNAs regulieren das Expressionsniveau von mRNAs durch den Prozess der RNA-Interferenz auf posttranskriptioneller Ebene hoch- oder herunter.10Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020.
kleine interferierende RNA (siRNA): 20-24 nt
kleine nukleolare RNA (snoRNA)
Piwi-interagierende RNA (piRNA): 24-31 nt. Sie interagieren mit Piwi-Proteinen der Argonaute-Familie und haben eine Funktion beim Targeting und der Spaltung von Transposons.11Cellerino & Sanguanini 2018, p. 12
Enhancer-RNA (eRNA) 12Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020.
Transkription zu Produktion eines RNA-Impfstoffes
Ein RNA-Impfstoff (auch: RNS-Impfstoff) beziehungsweise mRNA-Impfstoff (meistens Messenger-RNA, auch Boten-RNA bzw. mRNA genannt, sowie Nukleosid-modifizierte mRNA aus synthetischer Herstellung[1]) ist ein Impfstoff, dessen Wirkmechanismus auf Ribonukleinsäure (RNA oder RNS) beruht. RNA-Impfstoffe gehören zu den genetischen Impfstoffen, da aus der RNA ein Protein hergestellt wird, das eine Immunreaktion auslöst.
Durch die Verpackung der Erbinformation in Lipid–Nanopartikel wird die Aufnahme der Impfstoff-RNA in die Zellen der geimpften Person erleichtert.13Paul-Ehrlich-Institut: Wie funktionieren mRNA-Impfstoffe und was sind ihre Vorteile?. Auf: pei.de vom 30. Juli 2020; zuletzt abgerufen am 4. Februar 2021 Nachdem diese in die Zellflüssigkeit einer Zelle des Patienten gelangt ist, setzt sie eine Translation in Gang, welche das gewünschte Protein erzeugt. Die neu gebildeten Proteine, bezeichnet als Antigene, werden anschließend dem Immunsystem des Impflings präsentiert, das dann eine schützende Immunantwort erzeugt. Das Immunsystem lernt auf diese Weise im Verlauf der Immunreaktion, selektiv Zellen zu bekämpfen, die solche Antigene auf ihrer Zelloberfläche tragen, wie beispielsweise durch Viren infizierte Wirtszellen oder auch Tumorzellen14Rein Verbeke, Ine Lentacker, Stefaan C. De Smedt, Heleen Dewitte: Three decades of messenger RNA vaccine development. In: Nano Today. Nr. 28, 2019, S. 100766, doi:10.1016/j.nantod.2019.100766., weil es bei einem späteren Kontakt der geimpften Person mit den Erreger (oder weiteren Tumorzellen) deren Antigene wiedererkennt und so das Virus beziehungsweise die Infektionskrankheit (oder die Tumorzellen) gezielt bekämpfen kann.15Paul-Ehrlich-Institut: Mit welchen Technologien werden humane Virus-Impfstoffe entwickelt? Abschnitt: RNA/DNA-Impfstoffe. Auf: pei.de vom 30. Juli 2020; zuletzt abgerufen am 4. Februar 2021 Der Patient wird also gegen diese Erreger oder Tumorzellen immun, die RNA des Impfstoffes selbst erzeugt jedoch keine Immunreaktion und wird nach kurzer Zeit in der Zelle wieder abgebaut.
RNA-Impfstoffe können gegen alle proteinbasierten Antigene entwickelt werden, da nach einer Impfung mit einem RNA-Impfstoff bei der Translation ein Protein nach der RNA-Vorlage gebildet wird. Die Proteine können beispielsweise von Viren, Bakterien oder Tumoren (Tumorantigen) abgeleitet sein.
Datei:Video mRNA-Impfstoffe gegen das Coronavirus.webmMediendatei abspielen
Wie mRNA-Impfstoffe gegen das Coronavirus funktionieren.
Am 2. Dezember 2020 wurde während der COVID-19-Pandemie weltweit erstmals ein mRNA-Impfstoff durch eine staatliche Regulierungsbehörde zugelassen: Die britische Zulassungsbehörde MHRA verkündete eine Notzulassung für den SARS-CoV-2-Impfstoff Tozinameran, welcher von Biontech und Pfizer entwickelt wurde.16Kampf gegen Infektionskrankheiten: Erster mRNA-Impfstoff: Diese Technologie könnte Covid-19 besiegen – und nicht nur das. Abgerufen am 2. Dezember 2020. Dieser gilt nach aktueller Studienlage als sehr wirksam, sicher und gut verträglich.17Fernando P. Polack et al.: Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. In: The New England Journal of Medicine. Dezember 2020, doi:10.1056/NEJMoa2034577. Es folgten Zulassungen in weiteren Ländern (siehe zugelassene Impfstoffe).
Wirksamkeit und Sicherheit von RNA-Impfstoffen werden bereits seit mehreren Jahren in klinischen Studien am Menschen in anderen Anwendungsgebieten (verschiedenen Krebsarten, diverse Infektionskrankheiten) untersucht, ferner ist eine große Zahl präklinischer Daten verfügbar.18Ugur Sahin et al.: Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. In: Nature. Band 547, Nr. 7662, Juli 2017, S. 222–226, doi:10.1038/nature23003.19Martin Alberer et al.: Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. In: The Lancet. Band 390, Nr. 10101, Juli 2017, S. 1511–1520, doi:10.1016/S0140-6736(17)31665-3.20Norbert Pardi et al.: Recent advances in mRNA vaccine technology. In: Current Opinion in Immunology. Band 65, August 2020, S. 14–20, doi:10.1016/j.coi.2020.01.008.21Norbert Pardi et al.: mRNA vaccines – a new era in vaccinology. In: Nature Reviews Drug Discovery. Band 17, April 2018, S. 261–279, doi:10.1038/nrd.2017.243.22Oliver Klein und Katja Belousova: Impfstoffe: Warum es keine Langzeit-Nebenwirkungen gibt. In: ZDF. 6. Januar 2021, abgerufen am 14. Januar 2021.
Umfang der Studie
Im menschlichen Genom werden etwa 5 % aller Gene in RNA transkribiert.23C Frith, Martin; Pheasant, Michael; S Mattick, John (2005). “Genomics: The amazing complexity of the human transcriptome”. European Journal of Human Genetics. 13 (8): 894–897. doi:10.1038/sj.ejhg.5201459. PMID 15970949. S2CID 2836126. Das Transkriptom besteht aus kodierender mRNA, die etwa 1-4 % seiner Gesamtheit ausmacht, und nicht-kodierenden RNAs, die den Rest des Genoms ausmachen und keine Proteine hervorbringen.24Berg JMTJ, Stryer L. Biochemistry . New York: W H Freeman, 2002 25Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet 2006;15 Spec No 1:R17–29Die Anzahl der nicht-protein-kodierenden Sequenzen steigt in komplexeren Organismen.26U. Adams, Jill (2008). “Transcriptome: Connecting the Genome to Gene Function”. Nature Education. 1 (1): 195.
Mehrere Faktoren machen es schwierig, den Inhalt des Transkriptoms zu bestimmen. Dazu gehören unter anderem alternatives Spleißen, RNA-Editierung und alternative Transkription.27U. Adams, Jill (2008). “Transcriptome: Connecting the Genome to Gene Function”. Nature Education. 1 (1): 195. Außerdem sind Transkriptomtechniken in der Lage, die Transkription zu erfassen, die in einer Probe zu einem bestimmten Zeitpunkt stattfindet, obwohl sich der Inhalt des Transkriptoms während der Differenzierung ändern kann.28Peralta, Mihaela (2012). “The Human Transcriptome: An Unfinished Story”. Genes. 3 (3): 344–360. doi:10.3390/genes3030344. PMC 3422666. PMID 22916334. Die Hauptziele der Transkriptomik sind die folgenden “alle Arten von Transkripten zu katalogisieren, einschließlich mRNAs, nicht-kodierende RNAs und kleine RNAs; die Transkriptionsstruktur von Genen zu bestimmen, in Bezug auf ihre Startstellen, 5′- und 3′-Enden, Spleißmuster und andere posttranskriptionelle Modifikationen; und die sich verändernden Expressionsniveaus jedes Transkripts während der Entwicklung und unter verschiedenen Bedingungen zu quantifizieren.”29Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
Der Begriff kann auf die Gesamtmenge der Transkripte in einem bestimmten Organismus oder auf die spezifische Teilmenge der Transkripte, die in einem bestimmten Zelltyp vorhanden sind, angewendet werden. Im Gegensatz zum Genom, das für eine bestimmte Zelllinie grob fixiert ist (Mutationen ausgenommen), kann das Transkriptom mit den äußeren Umweltbedingungen variieren. Da es alle mRNA-Transkripte in der Zelle umfasst, spiegelt das Transkriptom die Gene wider, die zu einem bestimmten Zeitpunkt aktiv exprimiert werden, mit Ausnahme von mRNA-Abbauphänomenen wie der Transkriptionsabschwächung. Die Studie der Transkriptomik (die Expressionsprofilierung, Spleißvariantenanalyse usw. umfasst) untersucht das Expressionsniveau von RNAs in einer bestimmten Zellpopulation, wobei der Schwerpunkt oft auf mRNA liegt, aber manchmal auch andere wie tRNAs und sRNAs einbezogen werden.
Methoden der Konstruktion
Hauptartikel: Verfahren der Transkriptomik
Transkriptomik ist die quantitative Wissenschaft, die die Zuordnung einer Liste von Zeichenfolgen (“Reads”) zu einem Objekt (“Transkripte” im Genom) umfasst. Um die Expressionsstärke zu berechnen, wird die Dichte der Reads, die jedem Objekt entsprechen, gezählt.30Cellerino & Sanguanini 2018, p. preface Ursprünglich wurden Transkriptome mithilfe von Expressed-Sequence-Tags-Bibliotheken und der Serial and Cap Analysis of Gene Expression (SAGE) analysiert und untersucht.
Derzeit sind die beiden wichtigsten Transkriptomtechniken DNA-Microarrays und RNA-Seq. Beide Techniken erfordern eine RNA-Isolierung durch RNA-Extraktionstechniken, gefolgt von ihrer Abtrennung von anderen zellulären Komponenten und der Anreicherung der mRNA.31Bryant S, Manning DL (1998). “Isolation of messenger RNA”. RNA Isolation and Characterization Protocols. Methods in Molecular Biology. 86. pp. 61–4. doi:10.1385/0-89603-494-1:61. ISBN 978-0-89603-494-5. PMID 9664454.32Chomczynski P, Sacchi N (April 1987). “Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction”. Analytical Biochemistry. 162 (1): 156–9. doi:10.1016/0003-2697(87)90021-2. PMID 2440339.
Es gibt zwei allgemeine Methoden zur Ableitung von Transkriptomsequenzen. Ein Ansatz kartiert Sequenzlesungen auf ein Referenzgenom, entweder des Organismus selbst (dessen Transkriptom untersucht wird) oder einer eng verwandten Art. Der andere Ansatz, die De-novo-Transkriptom-Assemblierung, verwendet Software, um Transkripte direkt aus kurzen Sequenz-Reads abzuleiten, und wird bei Organismen mit nicht sequenzierten Genomen verwendet33Tachibana, Chris (31 July 2015). “Transcriptomics today: Microarrays, RNA-seq, and more”. Science Magazine. 349 (6247): 544. Bibcode:2015Sci…349..544T. Retrieved 2 May 2020..
DNA-Mikroarrays
Hauptartikel: DNA-Mikroarray
Die ersten Transkriptom-Studien basierten auf Microarray-Techniken (auch als DNA-Chips bekannt). Microarrays bestehen aus dünnen Glasschichten mit Spots, auf denen Oligonukleotide, so genannte “Sonden”, angeordnet sind; jeder Spot enthält eine bekannte DNA-Sequenz.34Schena, M.; Shalon, D.; Davis, R. W.; Brown, P. O. (20 October 1995). “Quantitative monitoring of gene expression patterns with a complementary DNA microarray”. Science. New York, N.Y.). 270 (5235): 467–470. Bibcode:1995Sci…270..467S. doi:10.1126/science.270.5235.467. ISSN 0036-8075. PMID 7569999. S2CID 6720459.
Bei der Durchführung von Microarray-Analysen wird mRNA von einer Kontroll- und einer Versuchsprobe gesammelt, wobei letztere in der Regel repräsentativ für eine Krankheit ist. Die RNA von Interesse wird in cDNA umgewandelt, um ihre Stabilität zu erhöhen, und mit Fluorophoren in zwei Farben, normalerweise grün und rot, für die beiden Gruppen markiert. Die cDNA wird auf der Oberfläche des Microarrays verteilt, wo sie mit den Oligonukleotiden auf dem Chip hybridisiert und mit einem Laser abgetastet wird. Die Fluoreszenzintensität an jedem Spot des Microarrays entspricht dem Niveau der Genexpression und anhand der Farbe der ausgewählten Fluorophore kann bestimmt werden, welche der Proben höhere Konzentrationen der interessierenden mRNA aufweist.35Govindarajan, Rajeshwar; Duraiyan, Jeyapradha; Kaliyappan, Karunakaran; Palanisamy, Murugesan (2012). “Microarray and its applications”. Journal of Pharmacy and Bioallied Sciences. 4 (6): S310-2. doi:10.4103/0975-7406.100283. PMC 3467903. PMID 23066278.

DNA-Mikroarray zum Nachweis der Genexpression in Proben von Mensch (links) und Maus (rechts)
Ein Microarray enthält in der Regel genügend Oligonukleotide, um alle bekannten Gene zu repräsentieren; die mit Microarrays gewonnenen Daten geben jedoch keine Auskunft über unbekannte Gene. In den 2010er Jahren wurden Microarrays fast vollständig durch Techniken der nächsten Generation ersetzt, die auf der DNA-Sequenzierung basieren.
RNA-Sequenzierung
Hauptartikel: RNA-Seq
Die RNA-Sequenzierung ist eine Sequenzierungstechnologie der nächsten Generation; als solche erfordert sie nur eine kleine Menge RNA und keine Vorkenntnisse des Genoms.36Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020. Sie ermöglicht sowohl eine qualitative als auch eine quantitative Analyse von RNA-Transkripten, wobei erstere die Entdeckung neuer Transkripte ermöglicht und letztere ein Maß für die relativen Mengen der Transkripte in einer Probe darstellt.37Cellerino & Sanguanini 2018, p. 12
Die drei Hauptschritte der Sequenzierung von Transkriptomen beliebiger biologischer Proben umfassen die RNA-Reinigung, die Synthese einer RNA- oder cDNA-Bibliothek und die Sequenzierung der Bibliothek.38Cellerino & Sanguanini 2018, p. 12 Der RNA-Reinigungsprozess unterscheidet sich für kurze und lange RNAs.39Cellerino & Sanguanini 2018, p. 12 Diesem Schritt folgt in der Regel eine Bewertung der RNA-Qualität mit dem Ziel, Verunreinigungen wie DNA oder technische Verunreinigungen im Zusammenhang mit der Probenverarbeitung zu vermeiden. Die RNA-Qualität wird mittels UV-Spektrometrie mit einem Absorptionspeak von 260 nm gemessen.40Cellerino & Sanguanini 2018, p. 13 Die RNA-Integrität kann auch quantitativ analysiert werden, indem das Verhältnis und die Intensität von 28S-RNA zu 18S-RNA verglichen werden, die im RNA-Integritäts-Score (RIN) angegeben werden.41Cellerino & Sanguanini 2018, p. 13 Da die mRNA die Spezies von Interesse ist und nur 3 % des Gesamtinhalts ausmacht, sollte die RNA-Probe behandelt werden, um rRNA und tRNA sowie gewebespezifische RNA-Transkripte zu entfernen.42Cellerino & Sanguanini 2018, p. 12
Der Schritt der Bibliothekspräparation mit dem Ziel, kurze cDNA-Fragmente herzustellen, beginnt mit der RNA-Fragmentierung auf Transkripte mit einer Länge zwischen 50 und 300 Basenpaaren. Die Fragmentierung kann enzymatisch (RNA-Endonukleasen), chemisch (Trismagnesiumsalzpuffer, chemische Hydrolyse) oder mechanisch (Sonikation, Vernebelung) erfolgen.43Cellerino & Sanguanini 2018, p. 18 Zur Umwandlung der RNA-Templates in cDNA wird die Reverse Transkription verwendet, wobei drei Priming-Methoden zur Anwendung kommen können, darunter die Oligo-DT, die Verwendung von Zufallsprimern oder die Ligierung spezieller Adaptor-Oligos.
Einzelzell-Transkriptomik
Hauptartikel: Einzelzell-Transkriptomik
Die Transkription kann auch auf der Ebene einzelner Zellen durch Einzelzell-Transkriptomik untersucht werden. Die Einzelzell-RNA-Sequenzierung (scRNA-seq) ist eine kürzlich entwickelte Technik, die die Analyse des Transkriptoms einzelner Zellen ermöglicht. Bei der Einzelzell-Transkriptomik werden auch Subpopulationen von Zelltypen berücksichtigt, die das interessierende Gewebe ausmachen.44Kanter, Itamar; Kalisky, Tomer (10 March 2015). “Single Cell Transcriptomics: Methods and Applications”. Frontiers in Oncology. 5: 53. doi:10.3389/fonc.2015.00053. ISSN 2234-943X. PMC 4354386. PMID 25806353. Mit diesem Ansatz lässt sich feststellen, ob Veränderungen in experimentellen Proben auf phänotypische zelluläre Veränderungen zurückzuführen sind, im Gegensatz zur Proliferation, bei der ein bestimmter Zelltyp in der Probe überexprimiert sein könnte. 45Stegle, Oliver; A. Teichmann, Sarah; C. Marioni, John (2015). “Computational and analytical challenges in single-cell transcriptomics”. Nature Reviews Genetics. 16 (3): 133–45. doi:10.1038/nrg3833. PMID 25628217. S2CID 205486032. Bei der Beurteilung der zellulären Progression durch Differenzierung sind durchschnittliche Expressionsprofile außerdem nur in der Lage, die Zellen nach der Zeit und nicht nach ihrem Entwicklungsstadium zu ordnen, und sind daher nicht in der Lage, Trends in den Genexpressionsniveaus aufzuzeigen, die für bestimmte Stadien spezifisch sind.46Trapnell, Cole (1 October 2015). “Defining cell types and states with single-cell genomics”. Genome Research. 25 (10): 1491–1498. doi:10.1101/gr.190595.115. ISSN 1088-9051. PMC 4579334. PMID 26430159. Einzelzell-Trarnscriptomic-Techniken wurden verwendet, um seltene Zellpopulationen wie zirkulierende Tumorzellen, Krebsstammzellen in soliden Tumoren und embryonale Stammzellen (ESCs) in Säugetier-Blastozysten zu charakterisieren.47Kanter, Itamar; Kalisky, Tomer (2015). “Single Cell Transcriptomics: Methods and Applications”. Frontiers in Oncology. 5 (13). doi:10.3389/fonc.2015.00053. PMC 4354386. PMID 25806353.
Obwohl es keine standardisierten Techniken für Einzelzell-Transkriptomik gibt, müssen mehrere Schritte durchgeführt werden. Der erste Schritt umfasst die Zellisolierung, die mit Niedrig- und Hochdurchsatztechniken durchgeführt werden kann. Es folgt ein qPCR-Schritt und dann die Einzelzell-RNAseq, bei der die RNA von Interesse in cDNA umgewandelt wird. Neuere Entwicklungen in der Einzelzell-Transkriptomik ermöglichen die Erhaltung des Gewebes und der subzellulären Lokalisation durch Kryo-Schneiden dünner Gewebeschnitte und Sequenzierung des Transkriptoms in jedem Schnitt. Eine andere Technik ermöglicht die Visualisierung einzelner Transkripte unter dem Mikroskop, wobei die räumliche Information jeder einzelnen Zelle, in der sie exprimiert werden, erhalten bleibt.48Kanter, Itamar; Kalisky, Tomer (2015). “Single Cell Transcriptomics: Methods and Applications”. Frontiers in Oncology. 5 (13). doi:10.3389/fonc.2015.00053. PMC 4354386. PMID 25806353.
Analyse
Eine Reihe von organismusspezifischen Transkriptom-Datenbanken wurden erstellt und annotiert, um die Identifizierung von Genen zu unterstützen, die in verschiedenen Zellpopulationen unterschiedlich exprimiert werden.
RNA-seq entwickelt sich (2013) zur Methode der Wahl für die Messung von Transkriptomen von Organismen, obwohl die ältere Technik der DNA-Mikroarrays immer noch verwendet wird.49Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660. RNA-seq misst die Transkription eines spezifischen Gens, indem lange RNAs in eine Bibliothek von cDNA-Fragmenten umgewandelt werden. Die cDNA-Fragmente werden dann mit Hilfe von Hochdurchsatz-Sequenzierungstechnologie sequenziert und an ein Referenzgenom oder Transkriptom angeglichen, das dann verwendet wird, um ein Expressionsprofil der Gene zu erstellen.50Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
Anwendungen
Säugetiere
Die Transkriptome von Stamm– und Krebszellen sind von besonderem Interesse für Forscher, die die Prozesse der zellulären Differenzierung und Karzinogenese verstehen wollen. Eine Pipeline, die RNA-seq- oder Genarray-Daten nutzt, kann verwendet werden, um genetische Veränderungen in Stamm- und Vorläuferzellen zu verfolgen und erfordert mindestens drei unabhängige Genexpressionsdaten vom erstgenannten Zelltyp und reifen Zellen.51Godoy, Patricio; Schmidt-Heck, Wolfgang; Hellwig, Birte; Nell, Patrick; Feuerborn, David; Rahnenführer, Jörg; Kattler, Kathrin; Walter, Jörn; Blüthgen, Nils; G. Hengstler, Jan (5 July 2018). “Assessment of stem cell differentiation based on genome-wide expression profiles”. Philosophical Transactions of the Royal Society B. 373 (1750): 20170221. doi:10.1098/rstb.2017.0221. PMC 5974444. PMID 29786556.
Die Analyse der Transkriptome menschlicher Eizellen und Embryonen wird verwendet, um die molekularen Mechanismen und Signalwege zu verstehen, die die frühe Embryonalentwicklung steuern, und könnte theoretisch ein mächtiges Werkzeug sein, um die richtige Auswahl der Embryonen bei der In-vitro-Fertilisation zu treffen. Analysen des Transkriptom-Inhalts der Plazenta im ersten Trimester der Schwangerschaft bei In-vitro-Fertilisation und Embryotransfer (IVT-ET) zeigten Unterschiede in der Genexpression, die mit einer höheren Häufigkeit von ungünstigen perinatalen Ergebnissen verbunden sind. Solche Erkenntnisse können genutzt werden, um die Praxis zu optimieren.52Zhao, L; Zheng, X; Liu, J; Zheng, R; Yang, R; Wang, Y; Sun, L (1 July 2019). “The placental transcriptome of the first-trimester placenta is affected by in vitro fertilization and embryo transfer”. Reproductive Biology and Endocrinology. 17 (1): 50. doi:10.1186/s12958-019-0494-7. PMC 6604150. PMID 31262321.Transkriptomanalysen können auch genutzt werden, um die Kryokonservierung von Eizellen zu optimieren, indem die mit dem Prozess verbundenen Verletzungen verringert werden.53Eroglu, Binnur; A. Szurek, Edyta; Schall, Peter; E. Latham, Keith; Eroglu, Ali (6 April 2020). “Probing lasting cryoinjuries to oocyte-embryo transcriptome”. PLOS ONE. 15 (4): e0231108. Bibcode:2020PLoSO..1531108E. doi:10.1371/journal.pone.0231108. PMC 7135251. PMID 32251418.
Die Transkriptomik ist ein aufstrebendes und stetig wachsendes Feld in der Entdeckung von Biomarkern, die bei der Beurteilung der Sicherheit von Medikamenten oder der Risikobewertung von Chemikalien eingesetzt werden können.54Szabo, David (2014). “Transcriptomic biomarkers in safety and risk assessment of chemicals”. Transcriptomic biomarkers in safety and risk assessment of chemicals. In Ramesh Gupta, editors:Gupta – Biomarkers in Toxicology, Oxford:Academic Press. pp. 1033–1038. doi:10.1016/B978-0-12-404630-6.00062-2. ISBN 978-0-12-404630-6.
Transkriptome können auch verwendet werden, um phylogenetische Beziehungen zwischen Individuen abzuleiten oder um evolutionäre Muster der Transkriptomerhaltung zu erkennen.55Drost, Hajk-Georg; Gabel, Alexander; Grosse, Ivo; Quint, Marcel; Grosse, Ivo (2018-05-01). “myTAI: evolutionary transcriptomics with R”. Bioinformatics. 34 (9): 1589–1590. doi:10.1093/molbev/msv012. ISSN 0737-4038. PMC 5925770. PMID 29309527.
Transkriptomanalysen wurden verwendet, um die Häufigkeit von Antisense-Transkription, ihre Rolle in der Genexpression durch Interaktion mit umgebenden Genen und ihre Häufigkeit in verschiedenen Chromosomen zu entdecken.56S, Katayama; et al. (2005). “Antisense Transcription in the Mammalian Transcriptome”. Science. 309 (5740): 1564–6. Bibcode:2005Sci…309.1564R. doi:10.1126/science.1112009. PMID 16141073. S2CID 34559885. RNA-seq wurde auch verwendet, um zu zeigen, wie RNA-Isoformen, Transkripte, die vom gleichen Gen stammen, aber unterschiedliche Strukturen haben, komplexe Phänotypen aus begrenzten Genomenen hervorbringen können.57Tachibana, Chris (31 July 2015). “Transcriptomics today: Microarrays, RNA-seq, and more”. Science Magazine. 349 (6247): 544. Bibcode:2015Sci…349..544T. Retrieved 2 May 2020.
Pflanzen
Transkriptomanalysen wurden genutzt, um die Evolution und den Diversifizierungsprozess von Pflanzenarten zu untersuchen. Im Jahr 2014 wurde das 1000 Plant Genomes Project abgeschlossen, in dem die Transkriptome von 1.124 Pflanzenarten aus den Familien Viridiplantae, Glaucophyta und Rhodophyta sequenziert wurden. Die Protein kodierenden Sequenzen wurden anschließend verglichen, um phylogenetische Beziehungen zwischen den Pflanzen abzuleiten und den Zeitpunkt ihrer Diversifizierung im Prozess der Evolution zu charakterisieren.58One Thousand Plant Transcriptomes Initiative (23 October 2019). “One thousand plant transcriptomes and the phylogenomics of green plants”. Nature. 574 (7780): 679–685. doi:10.1038/s41586-019-1693-2. PMC 6872490. PMID 31645766. Transkriptomstudien wurden genutzt, um die Genexpression in reifen Pollen zu charakterisieren und zu quantifizieren. Es wurde festgestellt, dass Gene, die am Zellwandstoffwechsel und am Zytoskelett beteiligt sind, überexprimiert werden. Transkriptom-Ansätze erlaubten es auch, Veränderungen in der Genexpression durch verschiedene Entwicklungsstadien des Pollens, von der Mikrospore bis zum reifen Pollenkorn, zu verfolgen; zusätzlich konnten solche Stadien über Arten verschiedener Pflanzen, einschließlich Arabidopsis, Reis und Tabak, verglichen werden.59Rutley, Nicholas; Twell, David (12 March 2015). “A decade of pollen transcriptomics”. Plant Reproduction. 28 (2): 73–89. doi:10.1007/s00497-015-0261-7. PMC 4432081. PMID 25761645.
Relation zu anderen ome-Feldern
Ähnlich wie bei anderen –om-basierten Technologien ermöglicht die Analyse des Transkriptoms einen unvoreingenommenen Ansatz bei der experimentellen Überprüfung von Hypothesen. Dieser Ansatz ermöglicht auch die Entdeckung von neuen Mediatoren in Signalwegen.60Cellerino & Sanguanini 2018, p. preface Wie bei anderen -omics basierten Technologien kann das Transkriptom im Rahmen eines Multiomics-Ansatzes analysiert werden. Es ist komplementär zu Metabolomics, aber im Gegensatz zu Proteomics kann keine direkte Assoziation zwischen einem Transkript und einem Metaboliten hergestellt werden.

Allgemeines Schema, das die Beziehungen zwischen dem Genom, Transkriptom, Proteom und Metabolom (Lipidom) zeigt.
Es gibt mehrere -ome Bereiche, die als Unterkategorien des Transkriptoms angesehen werden können. Das Exom unterscheidet sich vom Transkriptom dadurch, dass es nur die RNA-Moleküle umfasst, die in einer bestimmten Zellpopulation gefunden werden, und beinhaltet in der Regel die Menge oder Konzentration jedes RNA-Moleküls zusätzlich zu den molekularen Identitäten. Zusätzlich unterscheidet sich das Transkritpom auch vom Translatom, welches die Menge der RNAs ist, die eine Translation durchlaufen.
Der Begriff Meiom wird in der funktionellen Genomik verwendet, um das meiotische Transkriptom oder die Menge der RNA-Transkripte zu beschreiben, die während des Prozesses der Meiose produziert werden.61Crismani, Wayne; Baumann, Ute; Sutton, Tim; Shirley, Neil; Webster, Tracie; Spangenberg, German; Langridge, Peter; A Able, Jason (2006). “Microarray expression analysis of meiosis and microsporogenesis in hexaploid bread wheat”. BMC Genomics. 7 (267): 267. doi:10.1186/1471-2164-7-267. PMC 1647286. PMID 17052357. Die Meiose ist ein Hauptmerkmal der sich sexuell fortpflanzenden Eukaryoten und beinhaltet die Paarung homologer Chromosomen, die Synapse und die Rekombination. Da die Meiose in den meisten Organismen in einem kurzen Zeitraum stattfindet, ist das meiotische Transkript-Profiling aufgrund der Herausforderung der Isolierung (oder Anreicherung) der meiotischen Zellen (Meiozyten) schwierig. Wie bei Transkriptomanalysen kann das Meiom auf der Ebene des gesamten Genoms mit Hilfe von groß angelegten transkriptomischen Techniken untersucht werden.62D. Bovill, William; Deveshwar, Priyanka; Kapoor, Sanjay; A. Able, Jason (2009). “Whole genome approaches to identify early meiotic gene candidates in cereals”. Functional & Integrative Genomics. 9 (2): 219–29. doi:10.1007/s10142-008-0097-4. PMID 18836753. S2CID 22854431.Das Meiom ist in Säugetier- und Hefesystemen gut charakterisiert worden und in Pflanzen etwas weniger umfangreich.63Deveshwar, Priyanka; D Bovill, William; Sharma, Rita; A Able, Jason; Kapoor, Sanjay (9 May 2011). “Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice”. BMC Plant Biology. 11 (78): 78. doi:10.1186/1471-2229-11-78. PMC 3112077. PMID 21554676.
Das Thanatotranskriptom besteht aus allen RNA-Transkripten, die in den inneren Organen eines toten Körpers 24-48 Stunden nach dem Tod weiter exprimiert werden oder neu zu exprimieren beginnen. Dazu gehören auch Gene, die nach der fetalen Entwicklung gehemmt werden. Wenn das Thanatotranskriptom mit dem Prozess des programmierten Zelltodes (Apoptose) in Verbindung steht, kann es als apoptotisches Thanatotranskriptom bezeichnet werden. Analysen des Thanatotranskriptoms werden in der Rechtsmedizin eingesetzt.64Javan, G. T.; Can, I.; Finley, S. J.; Soni, S (2015). “The apoptotic thanatotranscriptome associated with the liver of cadavers”. Forensic Science, Medicine, and Pathology. 11 (4): 509–516. doi:10.1007/s12024-015-9704-6. PMID 26318598. S2CID 21583165.
Die eQTL-Kartierung kann genutzt werden, um die Genomik mit der Transkriptomik zu ergänzen; genetische Varianten auf DNA-Ebene und Genexpressionsmessungen auf RNA-Ebene.65Manzoni, Claudia; A Kia, Demis; Vandrovcova, Jana; Hardy, John; W Wood, Nicholas; A Lewis, Patrick; Ferrari, Raffaele (March 2018). “Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences”. Briefings in Bioinformatics. 19 (2): 286–302. doi:10.1093/bib/bbw114. PMC 6018996. PMID 27881428.
Beziehung zum Proteom
Weitere Informationen: Proteom
Das Transkriptom kann als eine Untermenge des Proteoms gesehen werden, d.h. der gesamten Menge an Proteinen, die von einem Genom exprimiert werden.
Die Analyse der relativen mRNA-Expressionsniveaus kann jedoch durch die Tatsache erschwert werden, dass relativ kleine Veränderungen in der mRNA-Expression große Veränderungen in der Gesamtmenge des entsprechenden Proteins in der Zelle hervorrufen können. Eine Analysemethode, bekannt als Gene Set Enrichment Analysis, identifiziert eher koregulierte Gennetzwerke als einzelne Gene, die in verschiedenen Zellpopulationen hoch- oder herunterreguliert sind.66Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545-50.
Obwohl Microarray-Studien die relativen Mengen verschiedener mRNAs in der Zelle aufzeigen können, sind die mRNA-Spiegel nicht direkt proportional zum Expressionsniveau der Proteine, für die sie kodieren.67Schwanhäusser, Björn; et al. (May 2011). “Global quantification of mammalian gene expression control” (PDF). Nature. 473 (7347): 337–342. Bibcode:2011Natur.473..337S. doi:10.1038/nature10098. PMID 21593866. S2CID 205224972.Die Anzahl der Proteinmoleküle, die unter Verwendung eines gegebenen mRNA-Moleküls als Vorlage synthetisiert werden, ist in hohem Maße von den Translationsinitiationsmerkmalen der mRNA-Sequenz abhängig; insbesondere die Fähigkeit der Translationsinitiationssequenz ist eine Schlüsseldeterminante bei der Rekrutierung der Ribosomen für die Proteinübersetzung.
Transkriptom Datenbanken
Siehe auch: Transkriptomik-Technologien § Transkriptom-Datenbanken
Ensembl: [2] OmicTools: [3] Transkriptom-Browser: [4] ArrayExpress: [5]
Siehe auch
Biologie Portal
Transkriptomik Technologien
Serielle Analyse der Genexpression
Liste der Omics-Themen in der Biologie
Metabolom
Genexpression
Gewichtete Gen-Koexpressions-Netzwerkanalyse
Funktionelle Genomik
Verweise
- Cellerino, A; Sanguanini, M (2018), Transcriptome Analysis: Introduction and Examples from the Neurosciences, doi:10.1007/978-88-7642-642-1, ISBN 978-88-7642-641-4
Weitere Lektüre
- Volker Erdmann, Jan Barciszewski: RNA Technologies, Springer Berlin, 2012, ISBN 978-3642264900.
- ^ Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545-50.
- ^ Laule O, Hirsch-Hoffmann M, Hruz T, Gruissem W, and P Zimmermann. (2006) Web-based analysis of the mouse transcriptome using Genevestigator. BMC Bioinformatics 7:311
- ^ Assou, S.; Boumela, I.; Haouzi, D.; Anahory, T.; Dechaud, H.; De Vos, J.; Hamamah, S. (2010). “Dynamic changes in gene expression during human early embryo development: From fundamental aspects to clinical applications”. Human Reproduction Update. 17 (2): 272–290. doi:10.1093/humupd/dmq036. PMC 3189516. PMID 20716614.
- ^ Ogorodnikov, A; Kargapolova, Y; Danckwardt, S. (2016). “Processing and transcriptome expansion at the mRNA 3′ end in health and disease: finding the right end”. Eur J Physiol. 468 (6): 993–1012. doi:10.1007/s00424-016-1828-3. PMC 4893057. PMID 27220521.
Der Text ist unter der Lizenz „Creative Commons Attribution/Share Alike“ verfügbar; Informationen zu den Urhebern und zum Lizenzstatus eingebundener Mediendateien (etwa Bilder oder Videos) können im Regelfall durch Anklicken dieser abgerufen werden. Möglicherweise unterliegen die Inhalte jeweils zusätzlichen Bedingungen.
Stand vom … “ → WpEn | → WpDeInfo-Krümel
Die Bewertungs-Funktion ist derzeit deaktiviert!
Materialien zur Verfügung gestellt durch: → WpEn | → WpDe
Inhalte wurden aus dem Englischen übersetzt und mit mit Teilen der deutschen Wikipedia kombiniert. Für Hyperlinks der übersetzten Texte, für die es keine deutschen Wiki-Einträge gab, wurden die englischen beibehalten.
Tooltips:
Die Texte in eventuell vorhandenen Tooltip-Fenstern wurden von der Redaktion des W3punkt.de bereitgestellt, sie entstammen in der Hauptsache den englisch- und deutschsprachigen Wikipedias.
Das Titelbild ist gemeinfrei und entstammt der Wikipedia.
Der Text ist unter der Lizenz „Creative Commons Attribution/Share Alike“ verfügbar;
Quellen und Tiefen
- 1Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
- 2Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020.
- 3GK, Sim; FC, Kafatos; CW, Jones; MD, Koehler; A, Efstratiadis; T., Maniatis (December 1979). “Use of a cDNA library for studies on evolution and developmental expression of the chorion multigene families”. Cell. 8 (4): 1303–16. doi:10.1016/0092-8674(79)90241-1. PMID 519770.
- 4Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020.
- 5Govindarajan, Rajeshwar; Duraiyan, Jeyapradha; Kaliyappan, Karunakaran; Palanisamy, Murugesan (2012). “Microarray and its applications”. Journal of Pharmacy and Bioallied Sciences. 4 (6): S310-2. doi:10.4103/0975-7406.100283. PMC 3467903. PMID 23066278.
- 6C Frith, Martin; Pheasant, Michael; S Mattick, John (2005). “Genomics: The amazing complexity of the human transcriptome”. European Journal of Human Genetics. 13 (8): 894–897. doi:10.1038/sj.ejhg.5201459. PMID 15970949. S2CID 2836126.
- 7Peralta, Mihaela (2012). “The Human Transcriptome: An Unfinished Story”. Genes. 3 (3): 344–360. doi:10.3390/genes3030344. PMC 3422666. PMID 22916334.
- 8Clancy, Suzanne (2008). “DNA Transcription”. Nature Education. 1 (11): 41.
- 9Clancy, Suzanne (2008). “DNA Transcription”. Nature Education. 1 (11): 41.
- 10Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020.
- 11Cellerino & Sanguanini 2018, p. 12
- 12Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020.
- 13Paul-Ehrlich-Institut: Wie funktionieren mRNA-Impfstoffe und was sind ihre Vorteile?. Auf: pei.de vom 30. Juli 2020; zuletzt abgerufen am 4. Februar 2021
- 14Rein Verbeke, Ine Lentacker, Stefaan C. De Smedt, Heleen Dewitte: Three decades of messenger RNA vaccine development. In: Nano Today. Nr. 28, 2019, S. 100766, doi:10.1016/j.nantod.2019.100766.
- 15Paul-Ehrlich-Institut: Mit welchen Technologien werden humane Virus-Impfstoffe entwickelt? Abschnitt: RNA/DNA-Impfstoffe. Auf: pei.de vom 30. Juli 2020; zuletzt abgerufen am 4. Februar 2021
- 16Kampf gegen Infektionskrankheiten: Erster mRNA-Impfstoff: Diese Technologie könnte Covid-19 besiegen – und nicht nur das. Abgerufen am 2. Dezember 2020.
- 17Fernando P. Polack et al.: Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. In: The New England Journal of Medicine. Dezember 2020, doi:10.1056/NEJMoa2034577.
- 18Ugur Sahin et al.: Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. In: Nature. Band 547, Nr. 7662, Juli 2017, S. 222–226, doi:10.1038/nature23003.
- 19Martin Alberer et al.: Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. In: The Lancet. Band 390, Nr. 10101, Juli 2017, S. 1511–1520, doi:10.1016/S0140-6736(17)31665-3.
- 20Norbert Pardi et al.: Recent advances in mRNA vaccine technology. In: Current Opinion in Immunology. Band 65, August 2020, S. 14–20, doi:10.1016/j.coi.2020.01.008.
- 21Norbert Pardi et al.: mRNA vaccines – a new era in vaccinology. In: Nature Reviews Drug Discovery. Band 17, April 2018, S. 261–279, doi:10.1038/nrd.2017.243.
- 22Oliver Klein und Katja Belousova: Impfstoffe: Warum es keine Langzeit-Nebenwirkungen gibt. In: ZDF. 6. Januar 2021, abgerufen am 14. Januar 2021.
- 23C Frith, Martin; Pheasant, Michael; S Mattick, John (2005). “Genomics: The amazing complexity of the human transcriptome”. European Journal of Human Genetics. 13 (8): 894–897. doi:10.1038/sj.ejhg.5201459. PMID 15970949. S2CID 2836126.
- 24Berg JMTJ, Stryer L. Biochemistry . New York: W H Freeman, 2002
- 25Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet 2006;15 Spec No 1:R17–29
- 26U. Adams, Jill (2008). “Transcriptome: Connecting the Genome to Gene Function”. Nature Education. 1 (1): 195.
- 27U. Adams, Jill (2008). “Transcriptome: Connecting the Genome to Gene Function”. Nature Education. 1 (1): 195.
- 28Peralta, Mihaela (2012). “The Human Transcriptome: An Unfinished Story”. Genes. 3 (3): 344–360. doi:10.3390/genes3030344. PMC 3422666. PMID 22916334.
- 29Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
- 30Cellerino & Sanguanini 2018, p. preface
- 31Bryant S, Manning DL (1998). “Isolation of messenger RNA”. RNA Isolation and Characterization Protocols. Methods in Molecular Biology. 86. pp. 61–4. doi:10.1385/0-89603-494-1:61. ISBN 978-0-89603-494-5. PMID 9664454.
- 32Chomczynski P, Sacchi N (April 1987). “Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction”. Analytical Biochemistry. 162 (1): 156–9. doi:10.1016/0003-2697(87)90021-2. PMID 2440339.
- 33Tachibana, Chris (31 July 2015). “Transcriptomics today: Microarrays, RNA-seq, and more”. Science Magazine. 349 (6247): 544. Bibcode:2015Sci…349..544T. Retrieved 2 May 2020.
- 34Schena, M.; Shalon, D.; Davis, R. W.; Brown, P. O. (20 October 1995). “Quantitative monitoring of gene expression patterns with a complementary DNA microarray”. Science. New York, N.Y.). 270 (5235): 467–470. Bibcode:1995Sci…270..467S. doi:10.1126/science.270.5235.467. ISSN 0036-8075. PMID 7569999. S2CID 6720459.
- 35Govindarajan, Rajeshwar; Duraiyan, Jeyapradha; Kaliyappan, Karunakaran; Palanisamy, Murugesan (2012). “Microarray and its applications”. Journal of Pharmacy and Bioallied Sciences. 4 (6): S310-2. doi:10.4103/0975-7406.100283. PMC 3467903. PMID 23066278.
- 36Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). “Chapter 4 – Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications”. Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. Retrieved 25 April 2020.
- 37Cellerino & Sanguanini 2018, p. 12
- 38Cellerino & Sanguanini 2018, p. 12
- 39Cellerino & Sanguanini 2018, p. 12
- 40Cellerino & Sanguanini 2018, p. 13
- 41Cellerino & Sanguanini 2018, p. 13
- 42Cellerino & Sanguanini 2018, p. 12
- 43Cellerino & Sanguanini 2018, p. 18
- 44Kanter, Itamar; Kalisky, Tomer (10 March 2015). “Single Cell Transcriptomics: Methods and Applications”. Frontiers in Oncology. 5: 53. doi:10.3389/fonc.2015.00053. ISSN 2234-943X. PMC 4354386. PMID 25806353.
- 45Stegle, Oliver; A. Teichmann, Sarah; C. Marioni, John (2015). “Computational and analytical challenges in single-cell transcriptomics”. Nature Reviews Genetics. 16 (3): 133–45. doi:10.1038/nrg3833. PMID 25628217. S2CID 205486032.
- 46Trapnell, Cole (1 October 2015). “Defining cell types and states with single-cell genomics”. Genome Research. 25 (10): 1491–1498. doi:10.1101/gr.190595.115. ISSN 1088-9051. PMC 4579334. PMID 26430159.
- 47Kanter, Itamar; Kalisky, Tomer (2015). “Single Cell Transcriptomics: Methods and Applications”. Frontiers in Oncology. 5 (13). doi:10.3389/fonc.2015.00053. PMC 4354386. PMID 25806353.
- 48Kanter, Itamar; Kalisky, Tomer (2015). “Single Cell Transcriptomics: Methods and Applications”. Frontiers in Oncology. 5 (13). doi:10.3389/fonc.2015.00053. PMC 4354386. PMID 25806353.
- 49Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
- 50Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). “RNA-Seq: a revolutionary tool for transcriptomics”. Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
- 51Godoy, Patricio; Schmidt-Heck, Wolfgang; Hellwig, Birte; Nell, Patrick; Feuerborn, David; Rahnenführer, Jörg; Kattler, Kathrin; Walter, Jörn; Blüthgen, Nils; G. Hengstler, Jan (5 July 2018). “Assessment of stem cell differentiation based on genome-wide expression profiles”. Philosophical Transactions of the Royal Society B. 373 (1750): 20170221. doi:10.1098/rstb.2017.0221. PMC 5974444. PMID 29786556.
- 52Zhao, L; Zheng, X; Liu, J; Zheng, R; Yang, R; Wang, Y; Sun, L (1 July 2019). “The placental transcriptome of the first-trimester placenta is affected by in vitro fertilization and embryo transfer”. Reproductive Biology and Endocrinology. 17 (1): 50. doi:10.1186/s12958-019-0494-7. PMC 6604150. PMID 31262321.
- 53Eroglu, Binnur; A. Szurek, Edyta; Schall, Peter; E. Latham, Keith; Eroglu, Ali (6 April 2020). “Probing lasting cryoinjuries to oocyte-embryo transcriptome”. PLOS ONE. 15 (4): e0231108. Bibcode:2020PLoSO..1531108E. doi:10.1371/journal.pone.0231108. PMC 7135251. PMID 32251418.
- 54Szabo, David (2014). “Transcriptomic biomarkers in safety and risk assessment of chemicals”. Transcriptomic biomarkers in safety and risk assessment of chemicals. In Ramesh Gupta, editors:Gupta – Biomarkers in Toxicology, Oxford:Academic Press. pp. 1033–1038. doi:10.1016/B978-0-12-404630-6.00062-2. ISBN 978-0-12-404630-6.
- 55Drost, Hajk-Georg; Gabel, Alexander; Grosse, Ivo; Quint, Marcel; Grosse, Ivo (2018-05-01). “myTAI: evolutionary transcriptomics with R”. Bioinformatics. 34 (9): 1589–1590. doi:10.1093/molbev/msv012. ISSN 0737-4038. PMC 5925770. PMID 29309527.
- 56S, Katayama; et al. (2005). “Antisense Transcription in the Mammalian Transcriptome”. Science. 309 (5740): 1564–6. Bibcode:2005Sci…309.1564R. doi:10.1126/science.1112009. PMID 16141073. S2CID 34559885.
- 57Tachibana, Chris (31 July 2015). “Transcriptomics today: Microarrays, RNA-seq, and more”. Science Magazine. 349 (6247): 544. Bibcode:2015Sci…349..544T. Retrieved 2 May 2020.
- 58One Thousand Plant Transcriptomes Initiative (23 October 2019). “One thousand plant transcriptomes and the phylogenomics of green plants”. Nature. 574 (7780): 679–685. doi:10.1038/s41586-019-1693-2. PMC 6872490. PMID 31645766.
- 59Rutley, Nicholas; Twell, David (12 March 2015). “A decade of pollen transcriptomics”. Plant Reproduction. 28 (2): 73–89. doi:10.1007/s00497-015-0261-7. PMC 4432081. PMID 25761645.
- 60Cellerino & Sanguanini 2018, p. preface
- 61Crismani, Wayne; Baumann, Ute; Sutton, Tim; Shirley, Neil; Webster, Tracie; Spangenberg, German; Langridge, Peter; A Able, Jason (2006). “Microarray expression analysis of meiosis and microsporogenesis in hexaploid bread wheat”. BMC Genomics. 7 (267): 267. doi:10.1186/1471-2164-7-267. PMC 1647286. PMID 17052357.
- 62
- 63Deveshwar, Priyanka; D Bovill, William; Sharma, Rita; A Able, Jason; Kapoor, Sanjay (9 May 2011). “Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice”. BMC Plant Biology. 11 (78): 78. doi:10.1186/1471-2229-11-78. PMC 3112077. PMID 21554676.
- 64
- 65Manzoni, Claudia; A Kia, Demis; Vandrovcova, Jana; Hardy, John; W Wood, Nicholas; A Lewis, Patrick; Ferrari, Raffaele (March 2018). “Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences”. Briefings in Bioinformatics. 19 (2): 286–302. doi:10.1093/bib/bbw114. PMC 6018996. PMID 27881428.
- 66Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545-50.
- 67Schwanhäusser, Björn; et al. (May 2011). “Global quantification of mammalian gene expression control” (PDF). Nature. 473 (7347): 337–342. Bibcode:2011Natur.473..337S. doi:10.1038/nature10098. PMID 21593866. S2CID 205224972.